Welcome to CINDERellA!
The goal of this project is to enable you to run causal Bayesian networks that accurately predict up and downstream genes and entire regulatory networks on the basis of gene expression or gene expression+genetics.
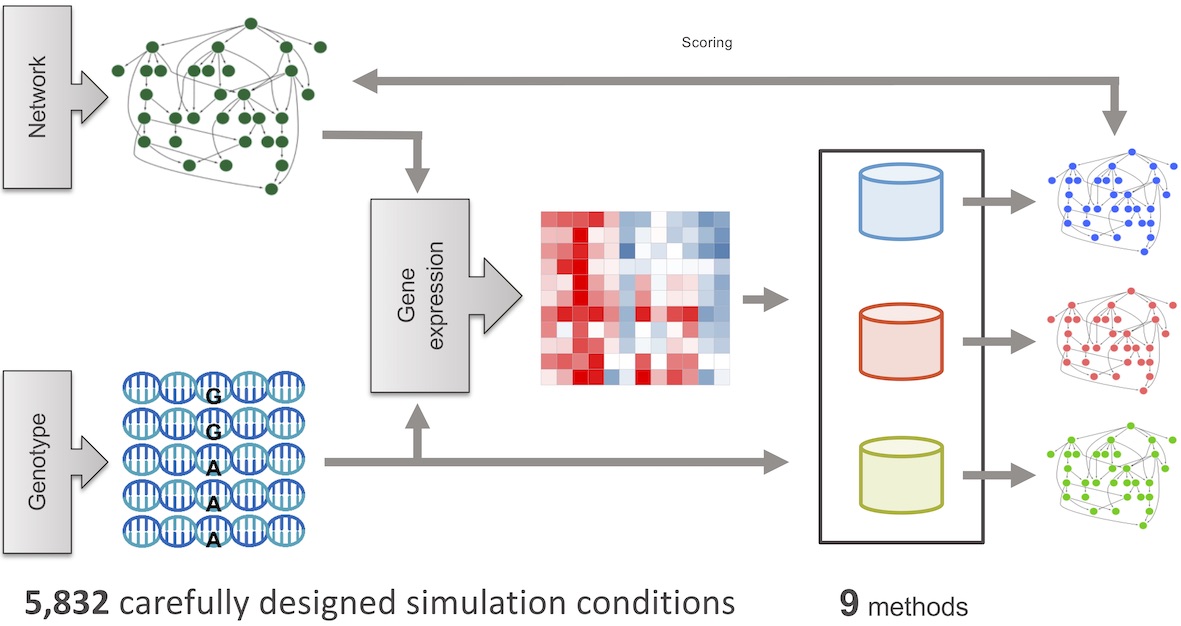
You can read more about methods included in this toolbox in this paper in Genetics where we compare their performance across ~14,000 realistic networks.
CINDERellA has been used to discover genes driving Alzheimer’s disease-related dementias (AD/ADRD) on the basis of bulk brain transcriptomes, brain multi-omics data 1, and brain multi-omics data 2.
Overview
CINDERellA is an easy-to-use Bayesian Network Learning Tool that learns causal networks from gene expression data using Markov Chain Monte Carlo (MCMC) methods.
⚠️ MATLAB Compatibility: This toolbox is compatible with MATLAB versions up to R2016a. Newer MATLAB versions may encounter compatibility issues.
Quick Start
Step 1: Setup
% Add CINDERellA to your MATLAB path
CINDERellA_PATH = './functions';
addpath(CINDERellA_PATH);
Step 2: Load Your Data
% Load expression data (user's responsibility - done outside the function)
expdata = read_exp('your_expression_data.txt');
Step 3: Run CINDERellA
% Basic usage with default parameters
CINDERellA(expdata.data);
Usage Examples
Basic Usage
% Load data first
expdata = read_exp('my_expression_data.txt');
% Run with default settings
CINDERellA(expdata.data);
Custom Parameters
% With custom parameters
CINDERellA(expdata.data, 'output_dir', 'my_results', ...
'max_parents', 3, 'runtime_minutes', 30, 'num_samples', 1000);
Using Prior Knowledge
% Create prior matrix to constrain search space
nGenes = size(expdata.data, 1);
prior = ones(nGenes, nGenes); % Start with all edges allowed
prior(1,2) = 0; % Disallow edge from gene 1 to gene 2
CINDERellA(expdata.data, 'prior_matrix', prior);
Visualization Options
% With layout and visualization options
CINDERellA(expdata.data, 'layout', 'force', 'edge_threshold', 0.33);
Input Parameters
Required
- data_matrix - Expression data matrix (rows=genes, columns=samples)
Optional Parameters
- ‘output_dir’ - Output directory (default:
'./CINDERellA_results/') - ‘sampler’ - MCMC sampler method (default:
'M.c2PB', or'M.REV50'if prior used) - ‘max_parents’ - Maximum parents per gene (default:
3) - ‘runtime_minutes’ - Total runtime in minutes (default:
0.5) - ‘num_samples’ - Number of network samples to collect (default:
100) - ‘edge_threshold’ - Edge frequency threshold for visualization (default:
0.3) - ‘prior_matrix’ - nGenes × nGenes binary matrix (1=allowed, 0=disallowed edges)
- ‘force_recompute’ - Force recomputation even if results exist (default:
false) - ‘layout’ - Network layout algorithm (default:
'force')
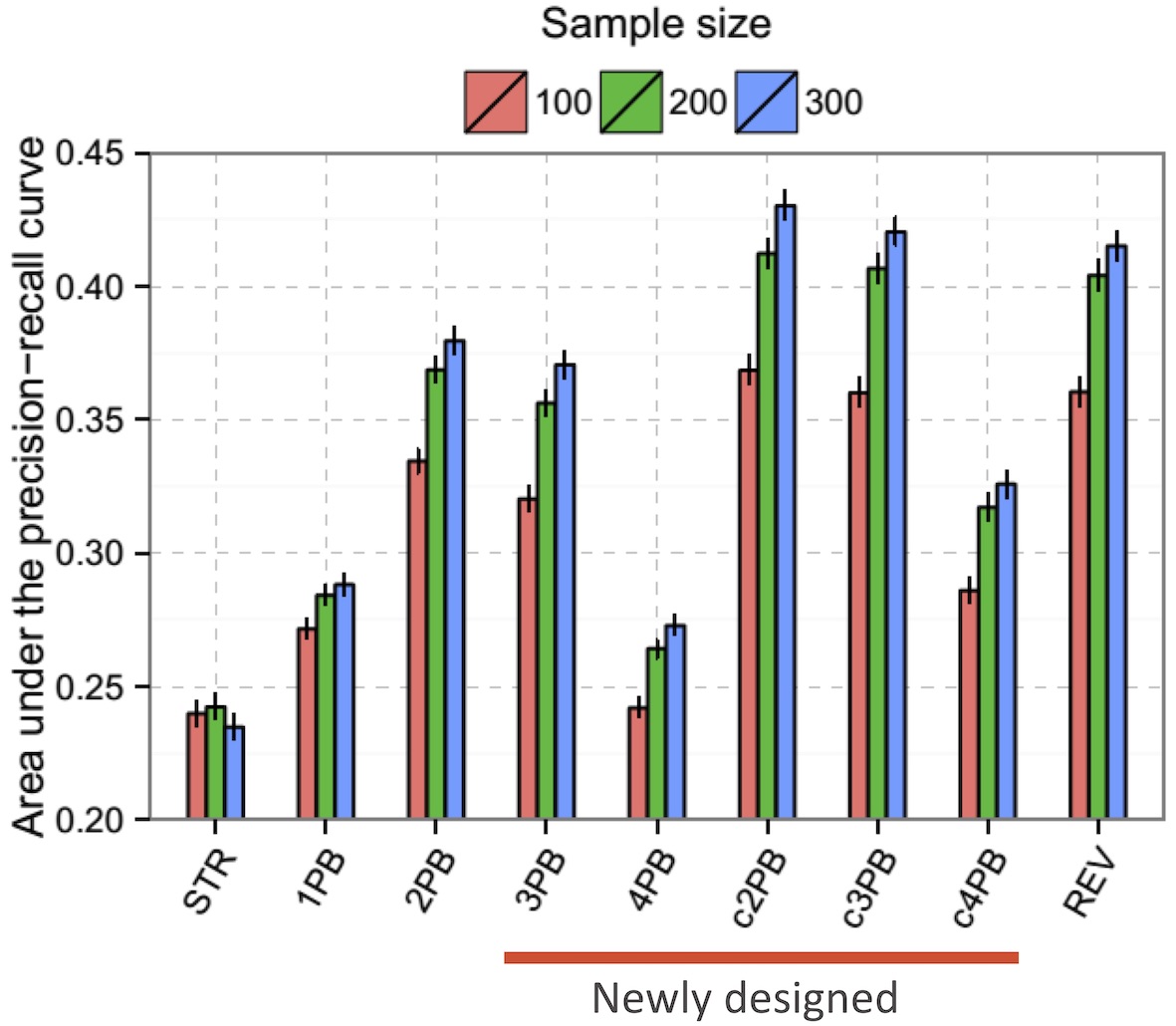
MCMC Samplers
Single Chain Samplers
'STR','c2PB','c3PB','c4PB','1PB','2PB','3PB','4PB','REV50'- Samples networks every (runtime/num_samples) seconds
Multi-Chain Samplers (Recommended)
'M.STR','M.c2PB','M.c3PB','M.c4PB','M.1PB','M.2PB','M.3PB','M.4PB','M.REV50'- Collects final network state from each chain after (runtime/num_samples) seconds
Layout Options
- ‘circle’ - Circular layout
- ‘force’ - Force-directed layout (default)
- ‘layered’ - Hierarchical layered layout
- ‘subspace’ - Subspace layout
Data Format Requirements
- Input file: Tab-separated text file
- Structure: Genes as rows, samples as columns
- Minimum: At least 2 genes and 2 samples
Output Files
CINDERellA generates several output files in the specified output directory:
- edgefrq.txt - Edge frequencies from sampled networks (main result for evaluation)
- Mcmc.mat - All sampled networks
- Param.mat - Parameters used in the analysis
- LS.mat - Local scores
- mcmc_diagnostics.png - Log likelihood trace plot
- network_visualization.png - Network plot with edges above threshold
Complete Workflow Example
% 1. Setup
CINDERellA_PATH = './functions';
addpath(CINDERellA_PATH);
% 2. Load data (user's responsibility)
expdata = read_exp('test_data/exp.txt');
% 3. Run CINDERellA
CINDERellA(expdata.data, 'runtime_minutes', 1, 'max_parents', 3);
% 4. Evaluation (done separately)
% Load true network if available
network = read_network('test_data/network.txt', size(expdata.data, 1));
% Load learned edge frequencies
edgefrq_data = dlmread('./CINDERellA_results/edgefrq.txt');
nGenes = size(expdata.data, 1);
edgefrq = sparse(edgefrq_data(:,1), edgefrq_data(:,2), edgefrq_data(:,3), nGenes, nGenes);
% Perform evaluation
[AUCPR, AUCROC] = evaluation(edgefrq, network.data, 'plot', 1);
Tips for Best Results
Runtime Settings
- Short test runs: 0.5-2 minutes for initial testing
- Production runs: 30-60 minutes for reliable results
- Complex networks: Consider longer runtimes for better convergence
Sampling Strategy
- Multi-chain samplers (M.* prefix) are generally recommended for final results
- Single chain samplers are useful for initial testing to see how much runtime is needed for convergence
Prior Knowledge
- Use
prior_matrixto incorporate known biological constraints - Set
prior(i,j) = 0to disallow edge from gene i to gene j - Set
prior(i,j) = 1to allow edge (default)
Visualization
- Adjust
edge_thresholdto control network complexity in plots - Higher thresholds show only strongest connections
- Lower thresholds show more potential connections
Troubleshooting
Common Issues
- Empty data matrix: Ensure your data file loaded correctly
- Dimension errors: Check that genes are rows, samples are columns
- Prior matrix size: Must be nGenes × nGenes
- Memory issues: Reduce
num_samplesorruntime_minutesfor large datasets
Performance Optimization
- Start with short runtime for testing
- Use appropriate number of samples (100-1000 typical)
- Consider your system’s computational capacity when setting parameters
Citation
If you use CINDERellA in your research, please cite:
Tasaki, S., Sauerwine, B., Hoff, B., Toyoshiba, H., Gaiteri, C., & Chaibub Neto, E. (2015). Bayesian network reconstruction using systems genetics data: comparison of MCMC methods. Genetics, 199(4), 973-989. doi:10.1534/genetics.114.172619
Author: Shinya Tasaki, Ph.D. (stasaki@gmail.com)
License: 3-clause BSD License